The aromatic hydrocarbons are known as arenes. Arenes are designated as Ar – H. The most characteristic reactions of arenes are substitution reactions. The electrophile displaces hydrogen atom and gets attached directly to the benzene ring. This is known as electrophilic aromatic substitution.

The electrophiles which take part in these substitution reactions are positively charged ions, electron deficient species with a partial positive charge and neutral molecules such as SO3. The five types of reactions, which comes under the category of electrophilic aromatic substitution are,
(i) Halogenation
(ii) Nitration
(iii) Sulphonation
(iv) Friedel–Crafts alkylation
(v) Friedel–Crafts acylation
General mechanism:
Benzene undergoes electrophilic substitution because of its exposed p electrons. In case of electrophilic substitution, benzene resembles alkene as the site of electrophilic attack in alkene is p bond only. But benzene also differs from alkene as the latter undergoes addition reaction and benzene prefers substitution rather than addition.
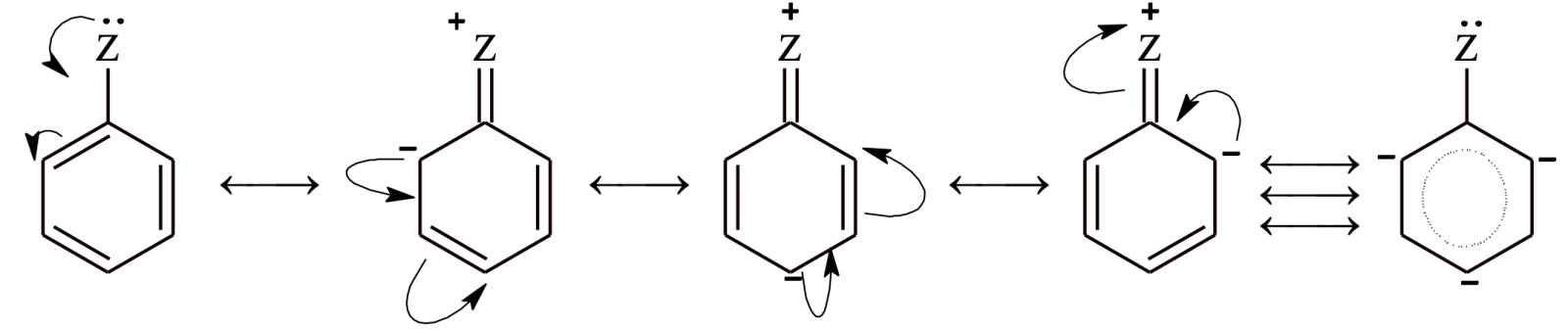
In step 1, the electrophile attacks the p system of benzene to form a non-aromatic cyclohexadienyl carbocation or arenium ion.
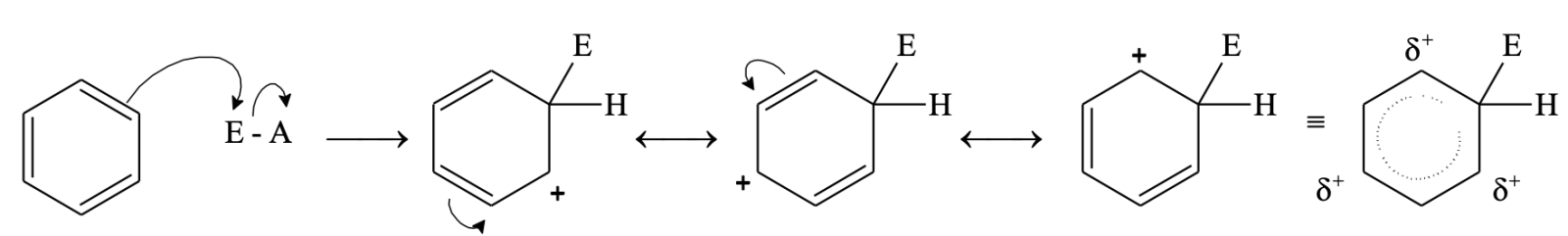
Now, the carbon atom attached to electrophile E is sp3 hybridized, so it does not have p-orbital available for delocalization. As a result, aromatic character is lost. The four p electrons are delocalized over 5 p-orbitals (delocalization is there but aromaticity is not there).
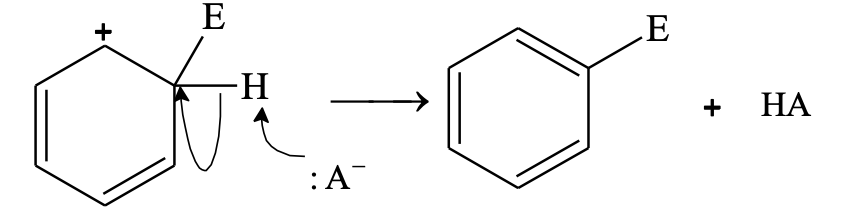
In step 2, a proton is removed from the carbon atom to which electrophile is attached. H+ leaves the ion leaving behind the shared pair of electron, which now forms a double bond and aromaticity is regained.
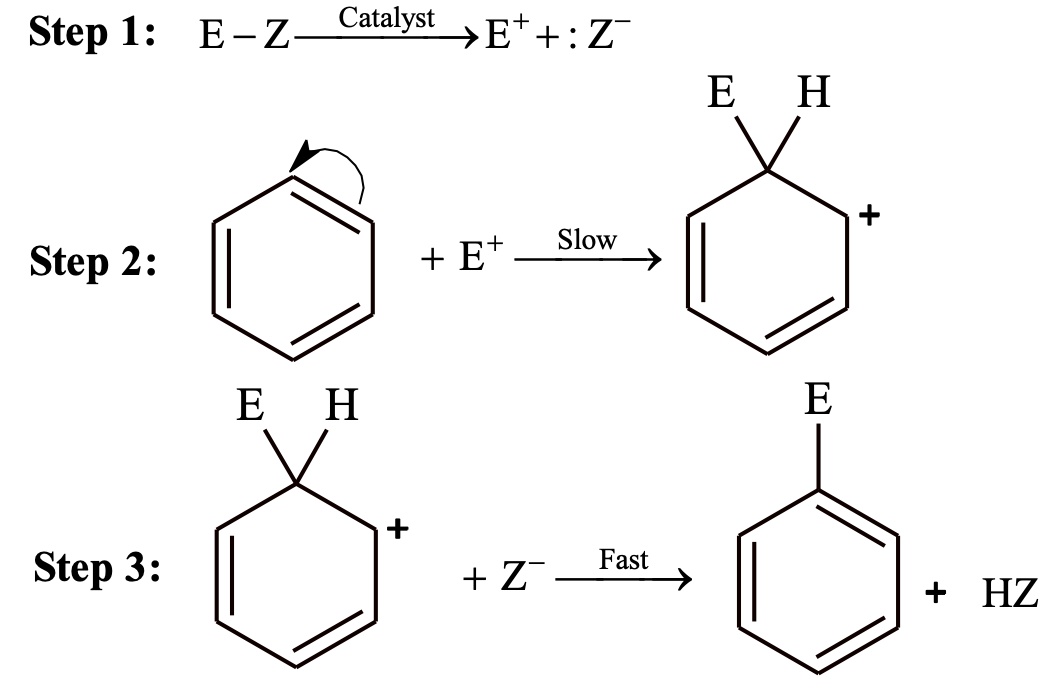
Halogenation
Direct halogenation of benzene is not possible and it does not decolourize bromine solution as there is no reaction. Benzene readily reacts with Br2 or Cl2 in presence of Lewis acids such as FeCl3, FeBr3, anhydrous AlCl3 etc.
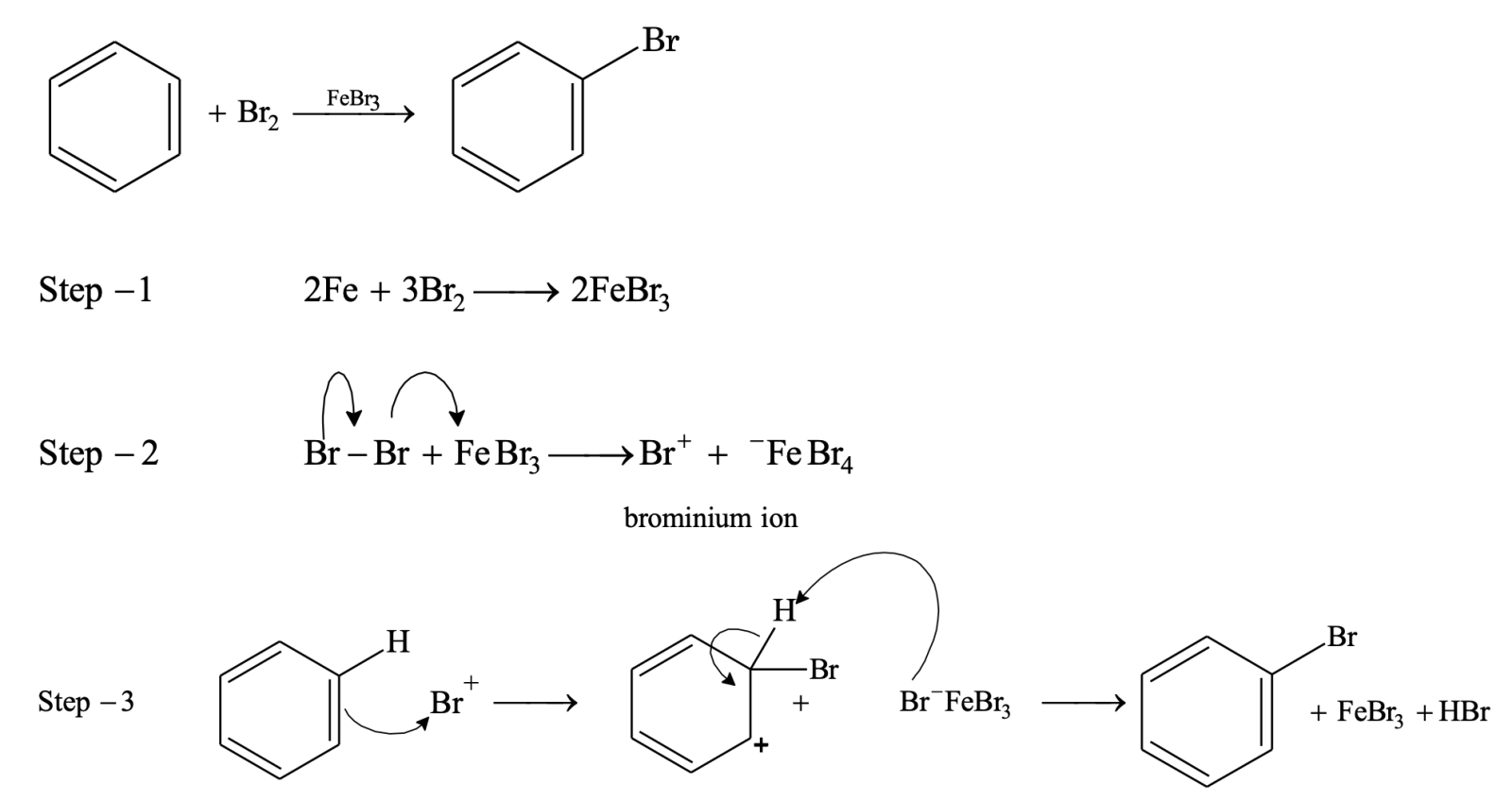
Fluorination takes place rapidly and always polyfluorinated benzene is formed rather than monofluorinated product. Iodine is unreactive towards halogenation of benzene.
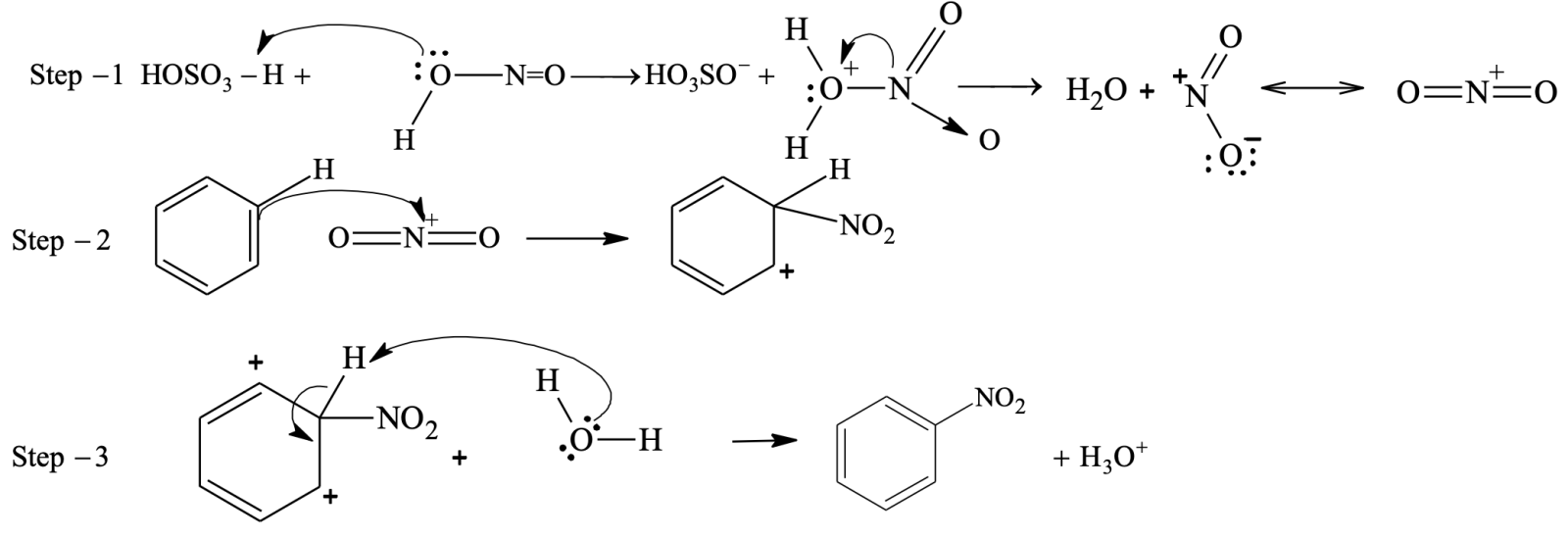
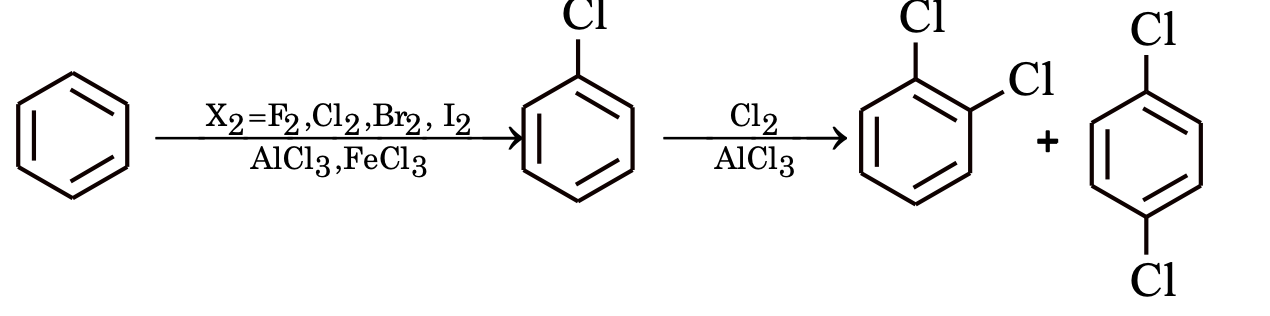
Nitration
Benzene reacts slowly with hot conc. HNO3 to yield nitrobenzene. The reaction is much faster if it is carried out in the presence of conc. H2SO4.
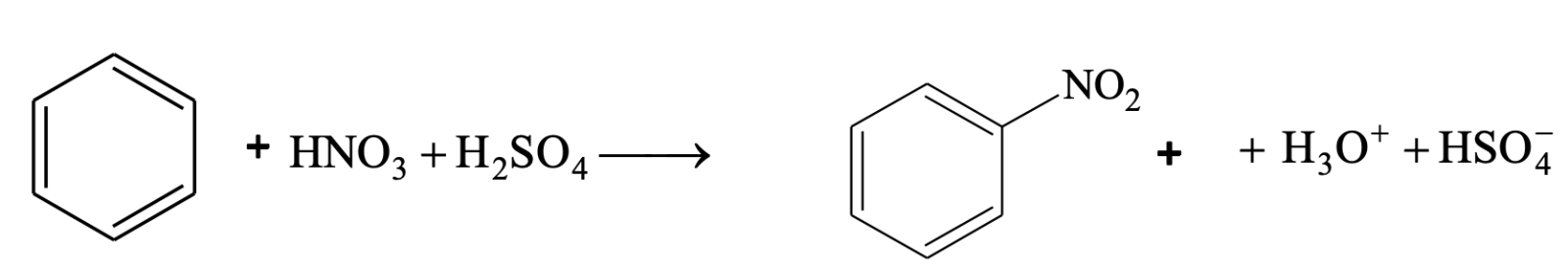
Here, H2SO4 acts as a an acid and HNO3 as a base.
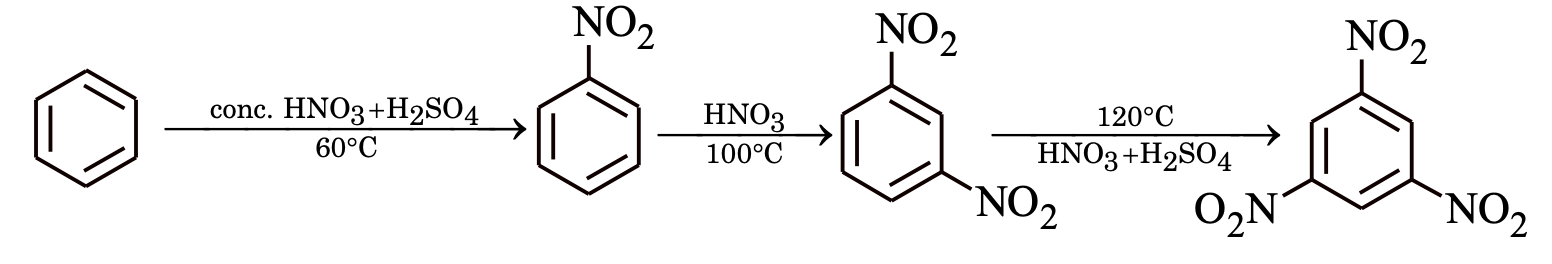
Sulphonation
Sulphonating agent is fuming H2SO4 + SO3 or conc. H2SO4.
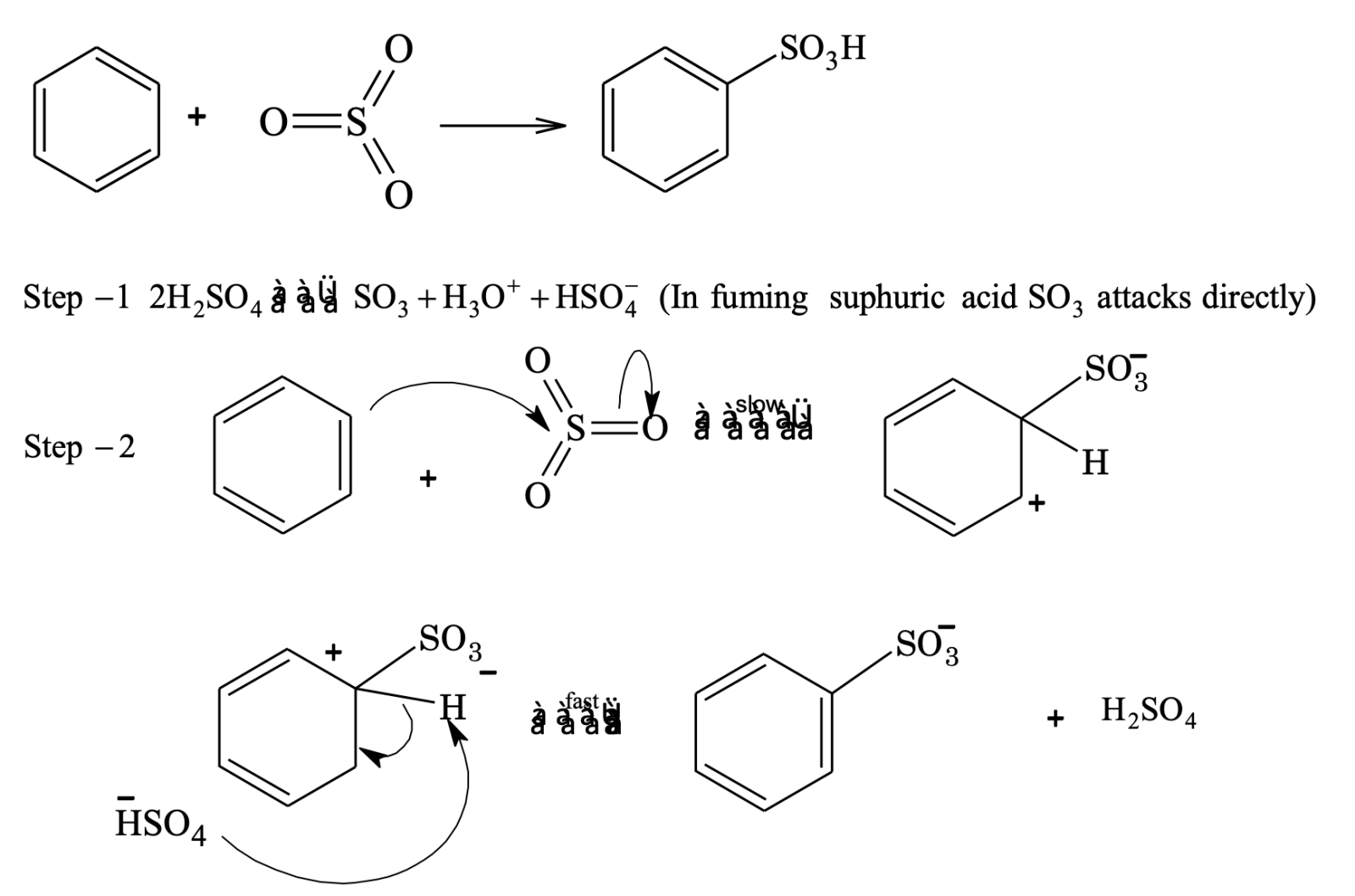
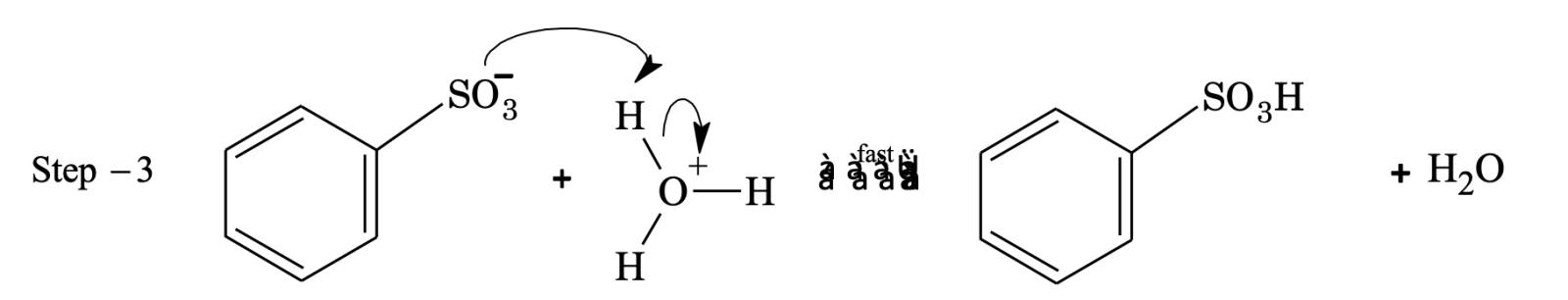
In sulphonation, all the steps are reversible.
To sulphonate benzene, concentrated H2SO4 acid is used (under these conditions position of equilibrium lies at the right).
To desulphonate benzene, dilute H2SO4 is used and steam is passed into the reaction mixture. Now the reaction equilibrium is shifted to left.
Friedel - Crafts Alkylation
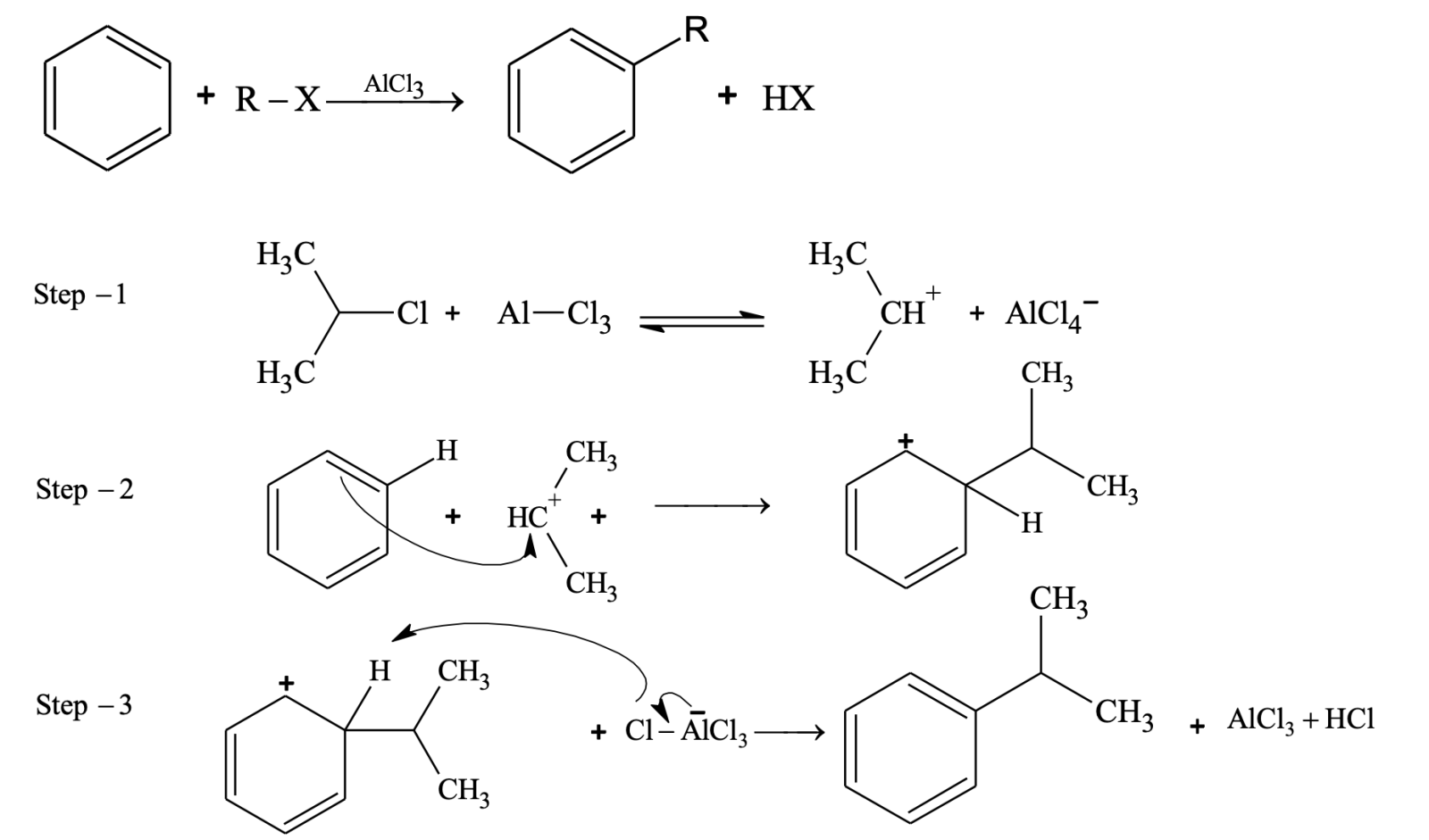
Other examples of alkylation:
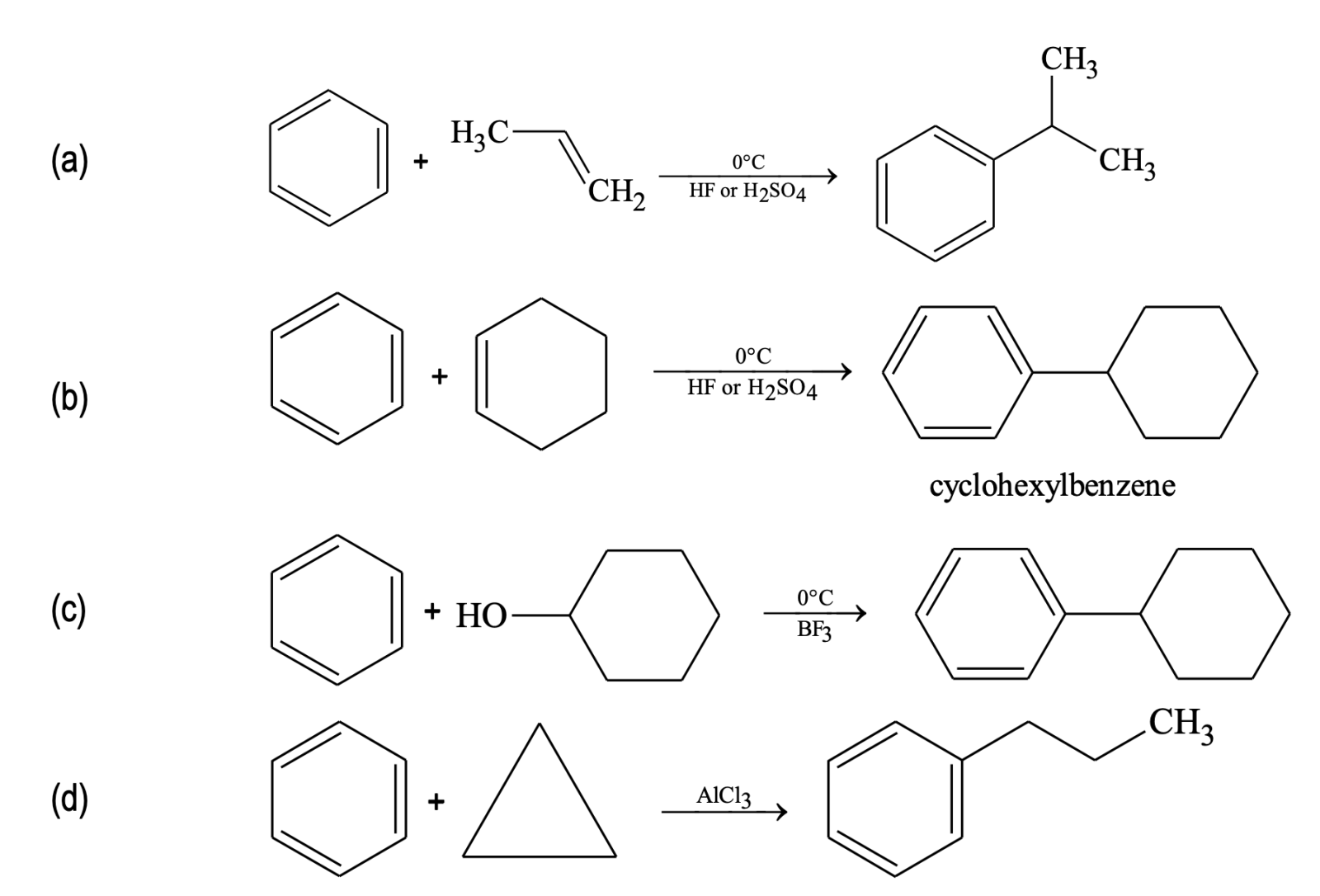
Friedel - Crafts Alkylation
Introduction of acyl group is called acylation (acyl group = RCO).
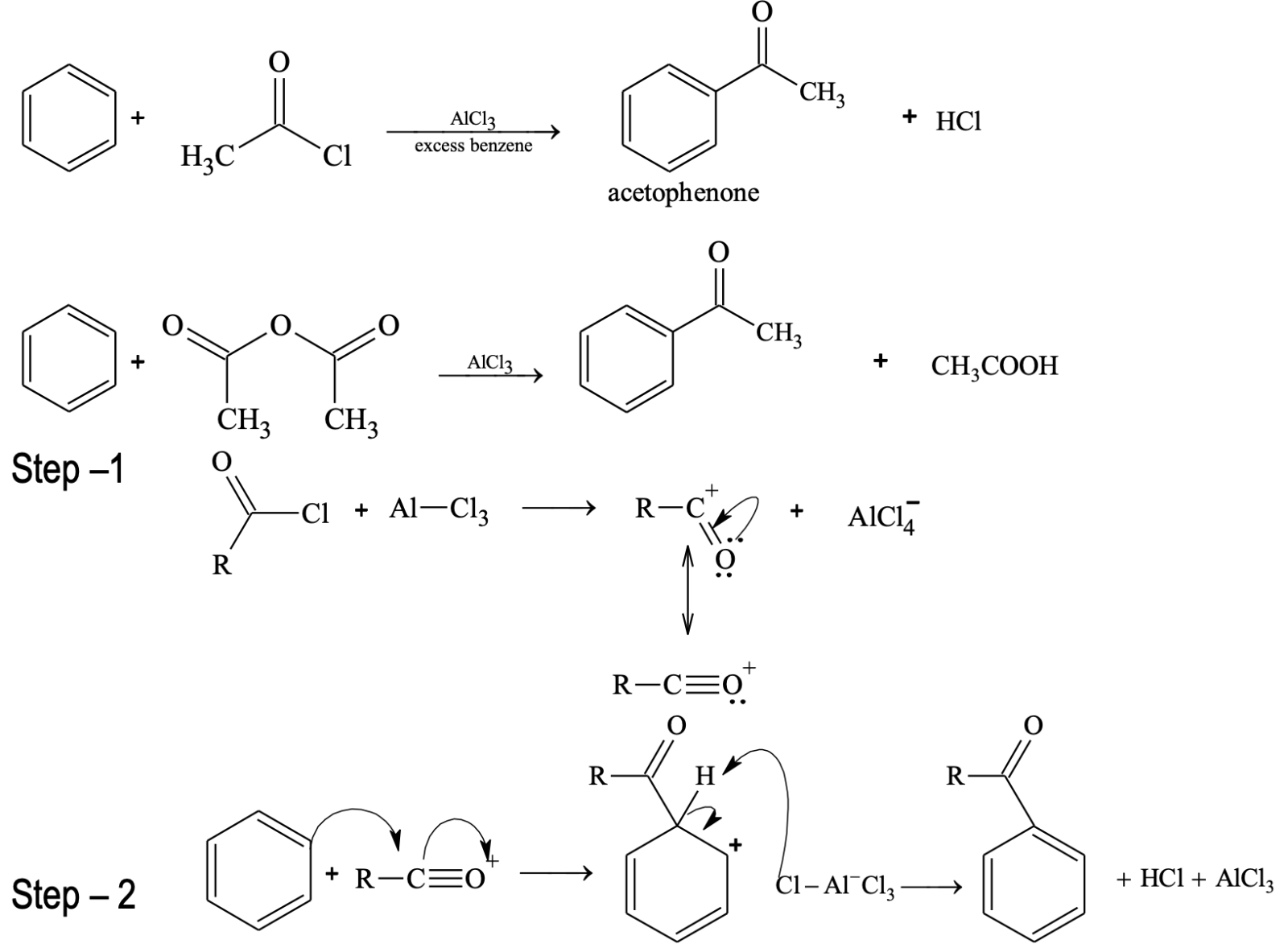
Limitations of Friedel - Crafts Reactions
- In alkylation carbocation can rearrange to more stable form (irrespective of whether it is formed from alkyl halide, alkene or alcohol).
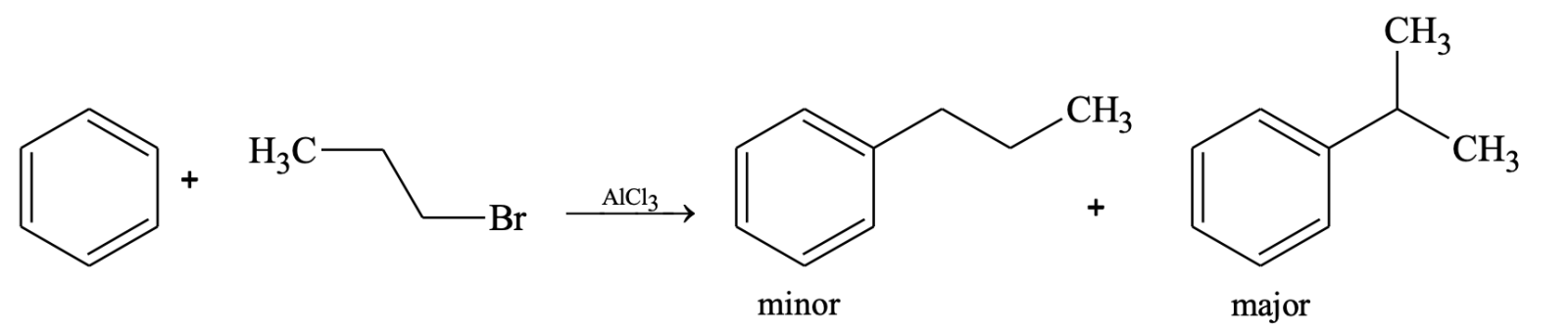
Rearrangements do not occur in Friedel–Crafts acylations. The acylium ion is resonance stabilized, so there is no need for it to rearrange.
- If the groups present are – NH2, -NHR, -NR2 or – OH, Friedel–Crafts reactions give very poor yields.
- Aryl and vinylic halides can’t be used as halide component because of the difficulty in producing the corresponding carbocations.
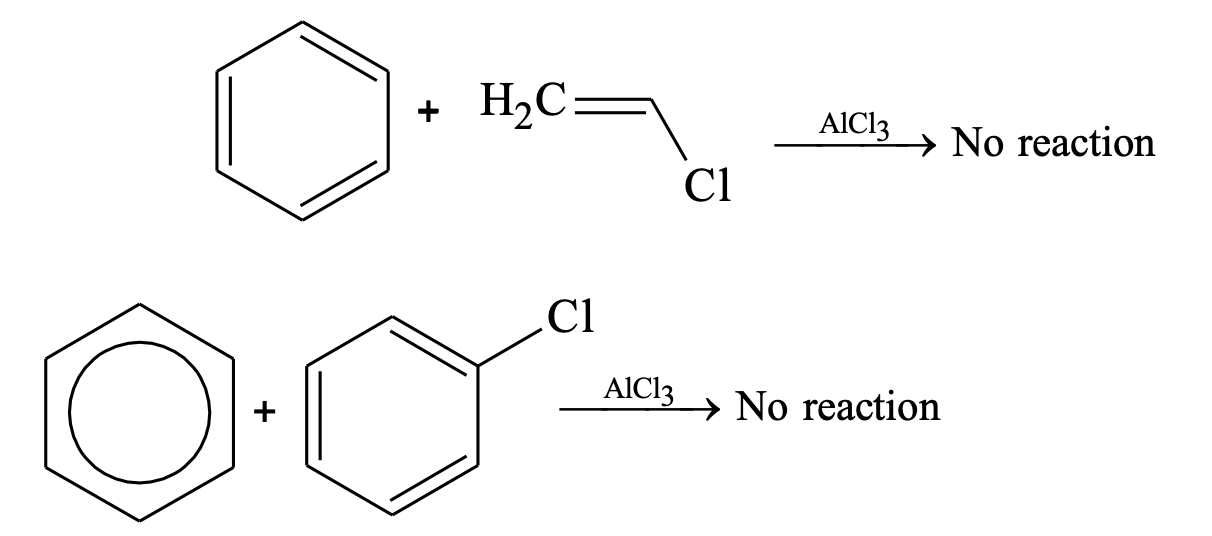
- Polyalkylations take place frequently during alkylation reactions but in case of acylation no such difficulty arises.
ORIENTATION AND REACTIVITY
In electrophilic aromatic substitutions, the substituents present in the benzene ring affects the rate of the reaction and the site of attack. So, the substituents affect both reactivity and orientation.
The substituent group can be activating or deactivating and the nature of groups is discussed in details with following examples.
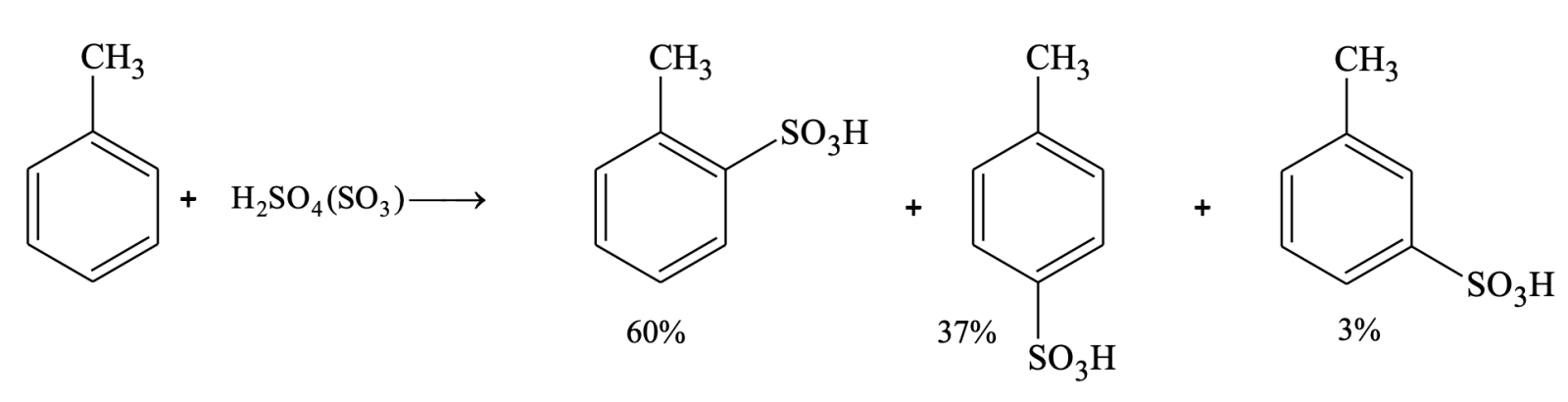
Electrophilic substitution of toluene is much faster in comparison to benzene. Sulphonation of benzene takes 20 to 30 minutes to complete whereas in case of toluene it is one minute only. Same results are obtained with nitration, halogenation and Friedel-Crafts reactions. From the above observation we can conclude that CH3 group activates the benzene ring towards electrophilic aromatic substitution. Like benzene , nitrobenzene also undergoes electrophilic substitution. Aniline is 106 times as reactive as benzene and nitro benzene is 1/106 as reactive as benzene.
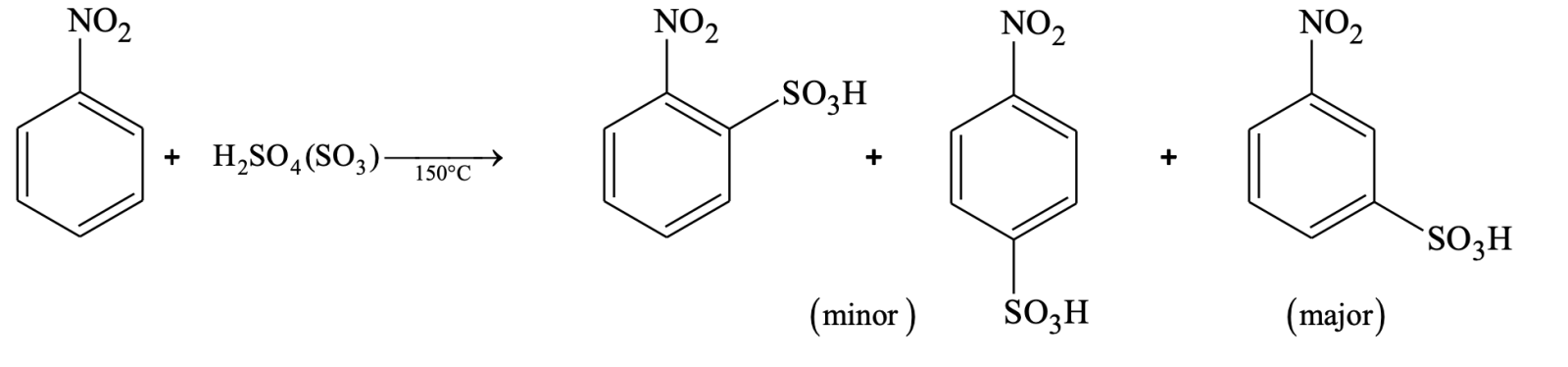
So, NO2 group deactivates the benzene ring towards electrophilic aromatic substitution. When the substituent is CH3 the products predominantly are p and o isomers. When the substituent is NO2, the major product is meta isomer. The groups which activate the benzene ring are called activating groups and those which deactivate the benzene ring are called deactivating groups.
Groups which direct the incoming group predominantly to o and p positions are called as o and p directors and those which direct predominantly to meta position are called meta directors.
Generally, activating groups are o and p directors and deactivating groups are m directors. But there is an exception, i.e. halogens. Halogens are deactivating groups but o and p directors.
Ortho and para directors:
Strongly activating
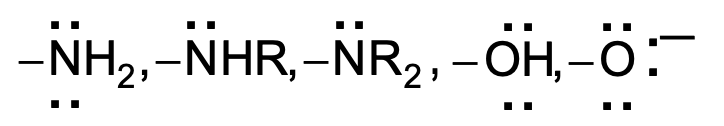
Moderately activating

Weakly activating
-CH3, - C2H5 – C6H5
Weakly deactivating
Meta directors:
- Moderately deactivating
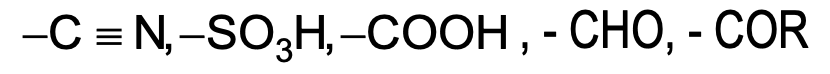
- Strongly deactivating
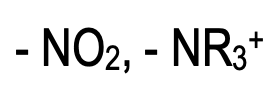
USE OF PROTECTING AND BLOCKING GROUPS
Very powerful activating groups such as amino and hydroxyl groups, make benzene ring so reactive that unwanted reactions take place. Some reagents used in electrophilic aromatic substitution such as HNO3 are also strong oxidizing agents. Both electrophilic and oxidizing agents seek electrons, so – OH and – NH2 group activates the ring not only towards electrophilic aromatic substitution but also towards oxidation.
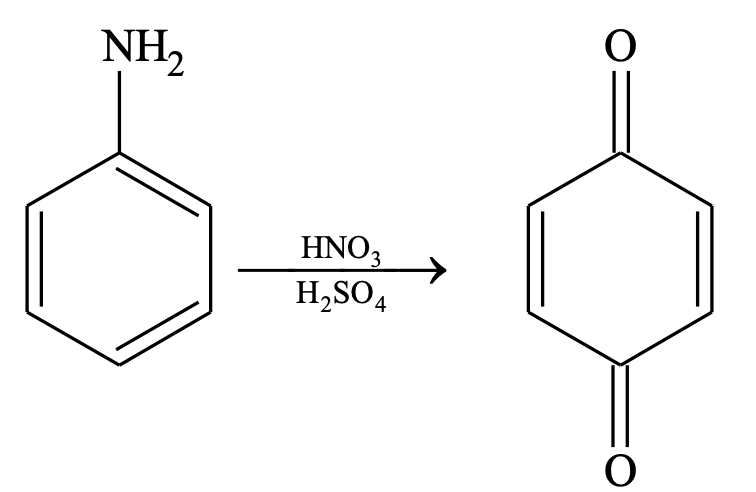
To avoid the oxidation of the substrate, blocking or protecting groups are used, like treating aniline with acetyl chloride or acetic anhydride converts aniline to acetanilide. Acetamide group is moderately activating and on nitration no oxidation takes place. After nitration is complete desired product can be obtained by removing the blocking group by hydrolysis.
Suppose, we want to prepare o-nitro aniline then we should follow the following procedure.
ORIENTATION (TWO SUBSTITUENTS)
(a) If one group is activating and the other group is deactivating, then the group which is activating in nature determines the orientation.
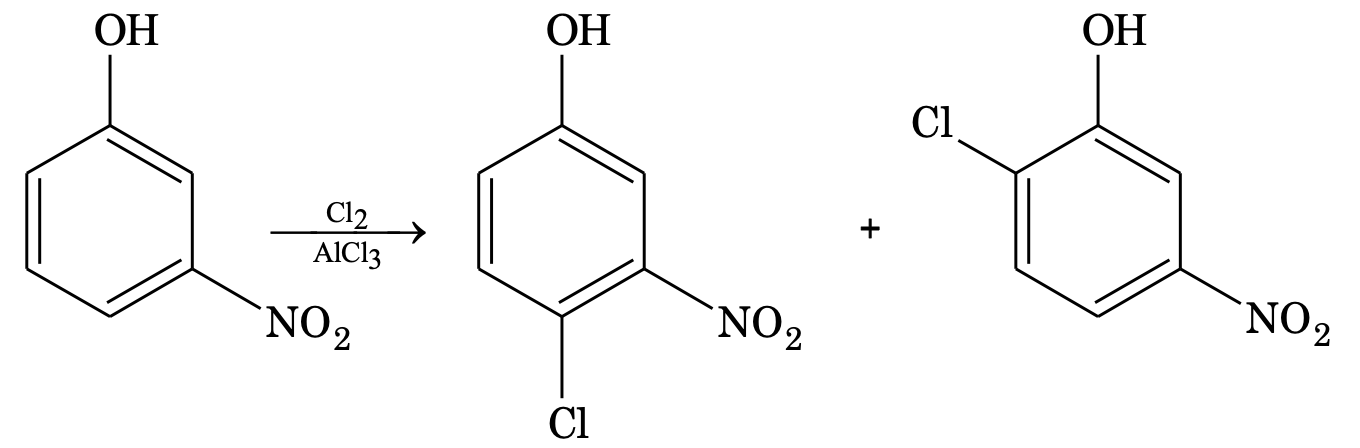
(b) If both are activating, the more activating group determines the orientation.
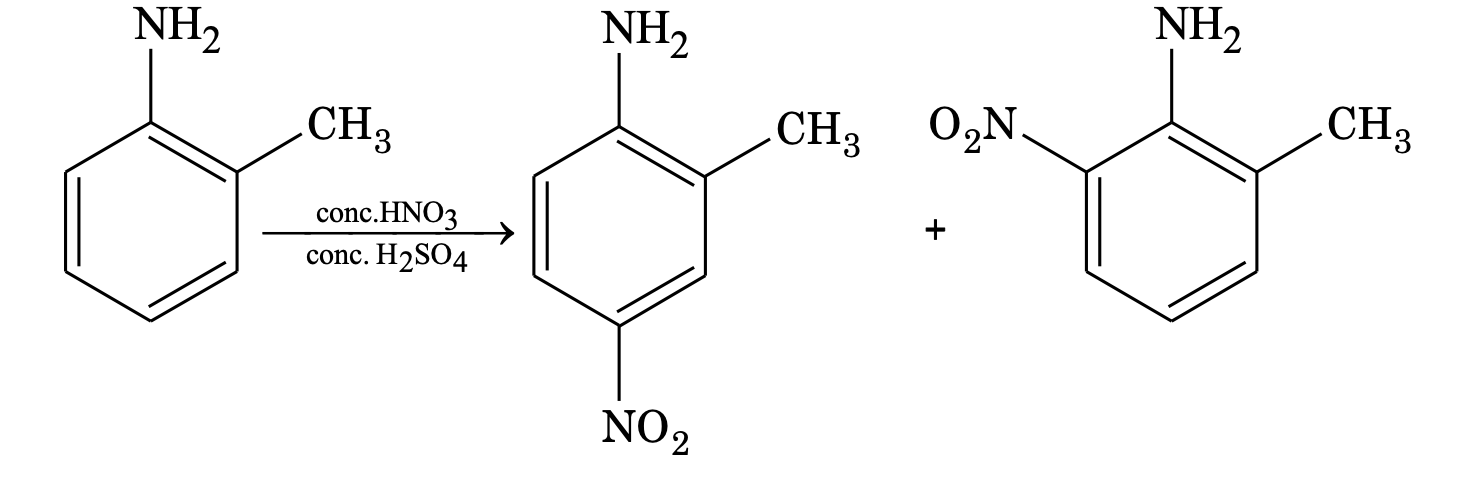
(c) If both are deactivating, less deactivating group determines the orientation
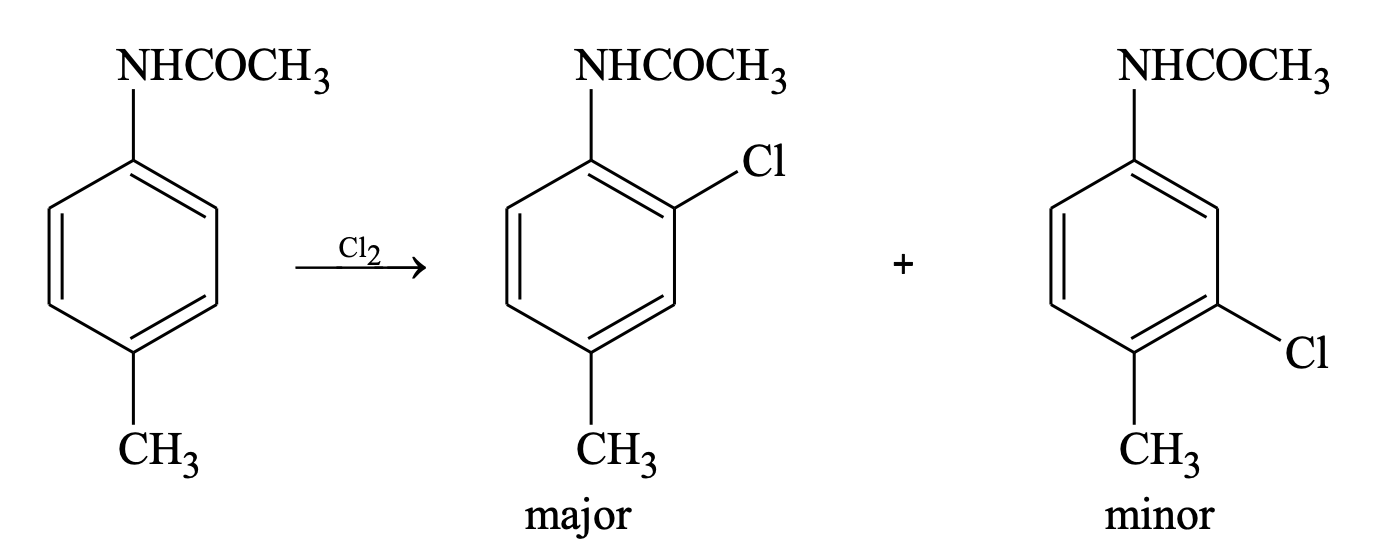
(d) If both groups are activating and reinforce at the same position, then there is no problem.
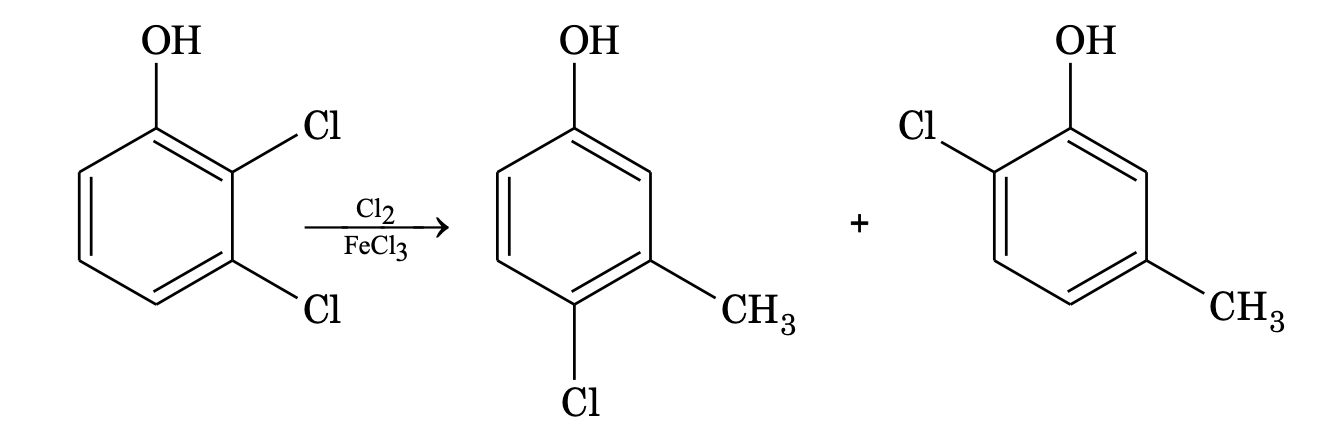
POINTS TO REMEBER
Aromatic Substitution Reaction
The replacement of one or more nuclear hydrogen atom by same number of substituents known as substitution reaction. These substitution reactions are halogenation, nitration, sulphonation, chloromethylation, formylation and Friedel–Crafts reaction.
General mechanism
Halogenation
Nitration
Sulphonation
Chloromethylation
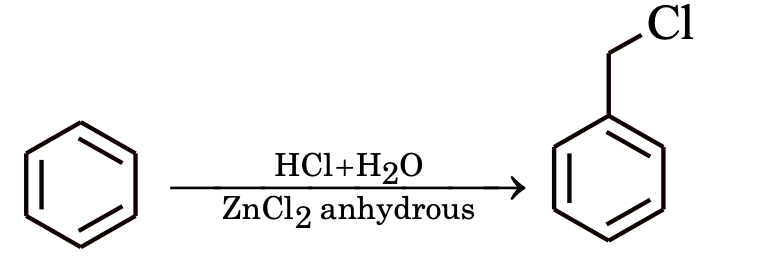
Formylation
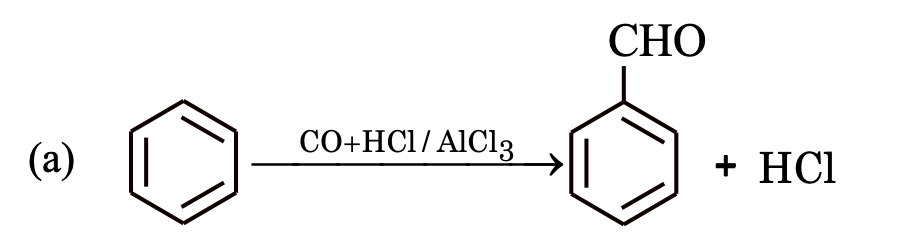
This reaction is known as Gattermann–Koch aldehyde synthesis.
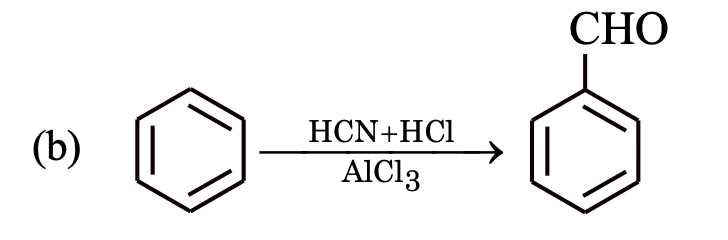
This reaction is known as Gattermann aldehyde reaction.
Friedel–Crafts Alkylation
Alkylation is possible with alkene, methanol and dialkyl sulphate.
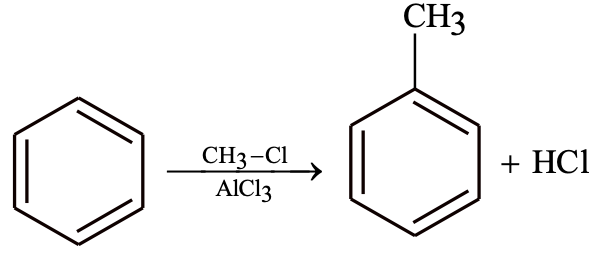
Friedel–Crafts Reactions
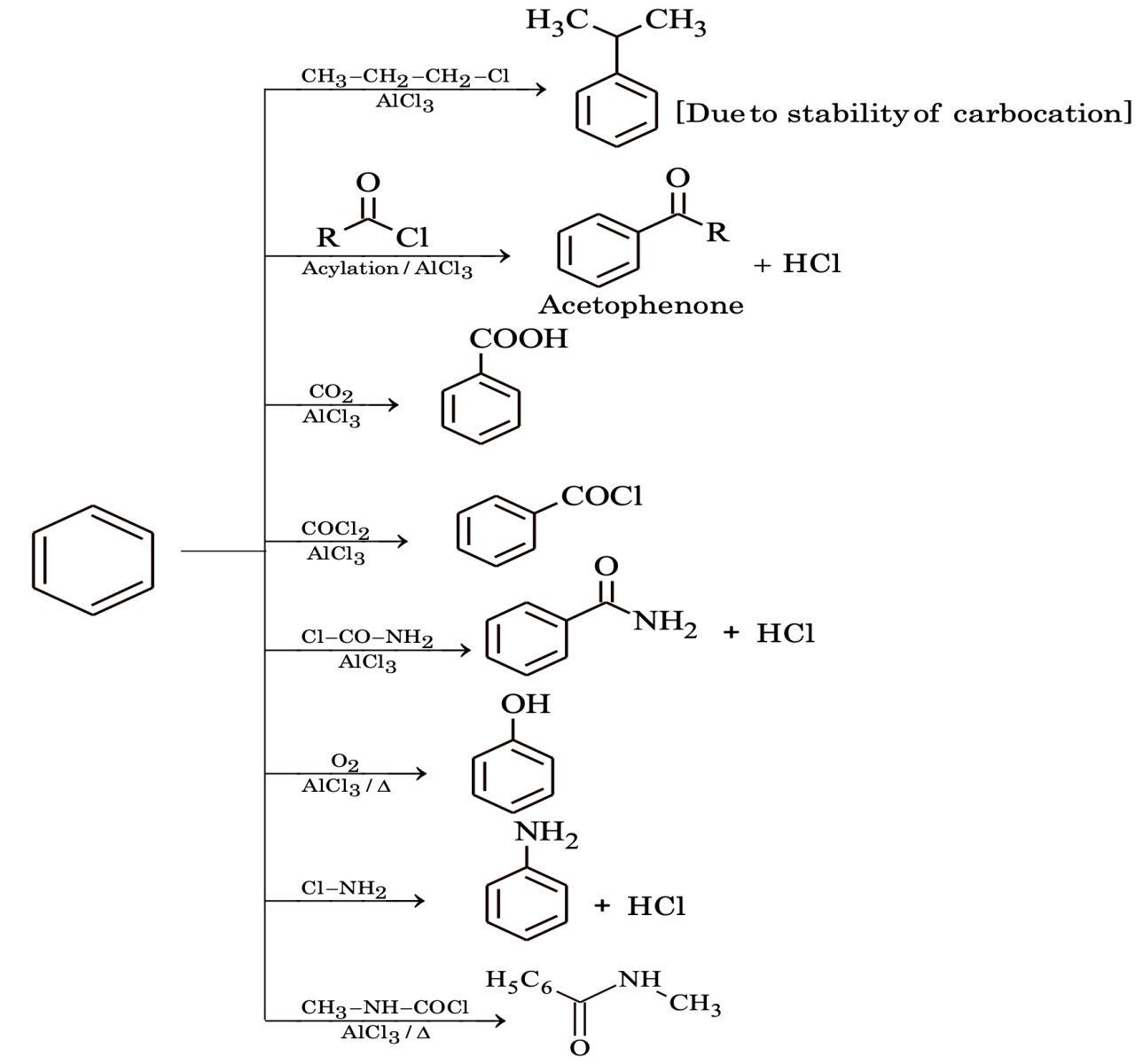
[Note: 1. Nitrobenzene do not undergoes Friedel–Crafts reaction due to deactivating nature of NO2 group.
- In aniline due to acid base reaction between NH2 and AlCl3 coordination bond will form and it behaves as a deactivating group.]
Directive Influence of Substituents
The group already present in a mono substituted product exerts a great influence on the position to be taken by the second incoming group. This is known as directive influence of the group.
Effect of substituents on electrophilic aromatic substitution
| Ortho and para directors | Meta directors |
|---|---|
| Strongly activating
−NH₂, −NHR, −NR₂, −OH, −O⁻
|
Moderately deactivating
−C≡N, −SO₃H, −COOH, −COOR, −CHO, −COR
|
| Moderately activating
−NHCOCH₃, −NHCOR, −OCH₃, −OR
|
Strongly deactivating
−NO₂, −NR₃⁺, −CF₃, −CCl₃
|
| Weakly activating
−CH₃, −C₂H₅, −R, −C₆H₅
|
|
| Weakly deactivating
−F, −Cl, −Br, −I
|
Key Points:
- Ortho and para directors: Direct incoming electrophiles to positions 2, 4, and 6 on the benzene ring
- Meta directors: Direct incoming electrophiles to positions 3 and 5 on the benzene ring
- Activating groups: Increase the rate of electrophilic aromatic substitution
- Deactivating groups: Decrease the rate of electrophilic aromatic substitution
1. When nitrobenzene is treated with Br2 in presence of FeBr3, the major product formed is m-bromonitrobenzene. Statements which are related to obtain m-isomer are
(A) the electron density on meta carbon is more than that on ortho and para positions.
(B) the intermediate carbonium ion formed after initial attack of Br+ at the meta position is least destabilized.
(C) loss of aromaticity when Br+ attacks at the ortho and para positions not at meta position.
(D easier loss of H+ to regain aromaticity from the meta position than from ortho and para positions.
Sol. (B)
2. The most basic compound among the following is
(A) benzylamine
(B) aniline
(C) acetanilide
(D) p-nitroaniline
Sol. (A)
Rest all show less tendency to donate electron pair due to resonance.
3. Toluene on oxidation with alkaline KMnO4 forms benzoic acid. What is the product formed when n-propyl benzene is oxidized with KMnO4?
(A) C6H5CH2COOH
(B) C6H5CH2CH2COOH
(C) C6H5COOH
(D) C6H5CHO
Sol. (C)
When any type of alkyl group (with benzylic hydrogen) is attached with benzene ring then this chain will be oxidized to carboxylic group.